Down syndrome
It is one of the most frequently occurring chromosomal abnormalities found in humans, occurring once in approximately every 800 to 1,000 live births.
John Langdon Down, an English physician, published an accurate description of a person with Down syndrome. It was this scholarly work, published in 1866, which earned Down the recognition as the “father” of the syndrome. Although other people had previously recognized the characteristics of the syndrome, it was Down who described the condition as a distinct and separate entity.
In 1959, the French physician, Jerome Lejeune, identified Down syndrome as a chromosomal anomaly. Instead of the usual 46 chromosomes present in each cell, Lejeune observed 47 in the cells of individuals with Down syndrome.

Down syndrome affects people of all races and economic levels. Women age 35 and older have a significantly increased risk of having a child with Down syndrome. A 35-year-old woman has a one in 400 chance of conceiving a child with Down syndrome, and this chance increases gradually to one in 110 by age 40. At age 45 the incidence becomes approximately one in 35.
Diagnosis
The diagnosis of Down syndrome is usually suspected after birth as a result of the baby’s appearance.
There are many physical characteristics which form the basis for suspecting an infant has Down syndrome. If Down syndrome is suspected, a karyotype will be performed to ascertain the diagnosis. Some infants with Down syndrome have only a few of these traits, while others have many.
Among the most common traits are:
- Muscle hypotonia, low muscle tone
- Flat facial profile, a somewhat depressed nasal bridge and a small nose.
- Oblique palpebral fissures, an upward slant to the eyes.
Dysplastic ear, an abnormal shape of the ear. Simian crease, a single deep crease across the centre of the palm.
- Hyper flexibility, an excessive ability to extend the joints
Dysplastic middle phalanx of the fifth finger, fifth finger has one flexion furrow instead of two.
- Epicanthal folds, small skin folds on the inner corner of the eyes.
- Excessive space between large and second toe.
- Enlargement of tongue in relationship to size of mouth.
All people with Down syndrome have some level of mental retardation. however, the level usually falls into the mild to moderate range .Children with Down syndrome learn to sit, walk, talk, play, toilet train and do most other activities, only somewhat later than others. Speech is often delayed.
Most people with Down syndrome have IQs that fall in the mild to moderate range of retardation. Children with Down syndrome are definitely educable
Today those with Down syndrome are working in various jobs and doing productive work. Banks, hotels, music, computer and many types of industries are employing individuals with Down syndrome.
People with Down syndrome date, socialize and form on-going relationships. Some are beginning to marry. Women with Down syndrome can and do have children, but there is a 50 percent chance that their child will have Down syndrome. Men with Down syndrome are believed to be sterile, with only one documented instance of a male with Down syndrome who has fathered a child.
Children with Down syndrome are at increased risk for certain health problems. Congenital heart defects, increased susceptibility to infection, respiratory problems, obstructed digestive tracts and childhood leukemia occur with greater frequency among children who have Down syndrome. However, advances in medicine have rendered most of these health problems treatable, and the majority of people born with Down syndrome today have a life expectancy of approximately fifty-five years.
Adults with Down syndrome are at increased risk for Alzheimer’s disease. Whereas approximately 6% of the general population will develop the disease, the figure is about 25% for people with Down syndrome.
Up to 50 percent of individuals with Down syndrome are born with congenital heart defects. The majority of heart defects in children with Down syndrome can now be surgically corrected with resulting long-term health improvements.
Leukemia – Individuals with Down syndrome have a 15 to 20 times greater risk of developing leukemia. The majority of cases are categorized as acute megakaryoblastic leukemia, which tends to occur in the first three years of life, and for which there is a high cure rate. A transient form of leukemia is also seen in newborns with Down syndrome, disappearing spontaneously during the first two to three months of life.
Screening tests for Down syndrome
There are two types of procedures available to pregnant women: Screening tests and diagnostic tests. Screening tests estimate the risk of the fetus having Down syndrome; diagnostic tests tell whether or not the fetus actually has the condition.
The most commonly used screening tests are the Triple Screen and the Alpha-fetoprotein Plus. These tests measure quantities of various substances in the blood (alpha-fetoprotein, human chorionic gonadotropin and unconjugated estriol) and together with the woman’s age, estimate her risk of having a child with Down syndrome. These screening tests are typically offered between fifteen and twenty weeks of gestation.
Screening tests are of limited value and are often performed in conjunction with a detailed sonogram. These tests are only able to accurately detect about sixty percent of fetuses with Down syndrome.
The procedures available for prenatal diagnosis of Down syndrome are chorionic villus sampling (CVS), amniocentesis and percutaneous umbilical blood sampling (PUBS). Each one of these procedures carries a small risk of miscarriage as tissue is extracted from the placenta or the umbilical cord to examine the fetus’s chromosomes. The procedures are about 98 to 99 percent accurate in the detection of Down syndrome.


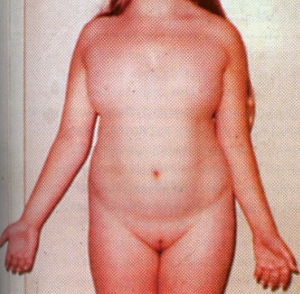 no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief.
no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief. The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.
The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.




