
Thalassaemia
Thalassaemia is inherent impairment of haemoglobin production due to partial or complete failure to synthesize a specific type of globin chain.
Hereditary disorders that result in a structurally abnormal hemoglobin or an insufficient quantity of hemoglobin are the most common genetic diseases of humans.
The production of hemoglobin requires a supply of iron, synthesis of heme, and synthesis of globin. Hemoglobin is essential for normal oxygen delivery by red blood cells.
When the condition is Heterozygous, the production of hemoglobin is mildly affected and the condition causes little disability. The synthesis is grossly affected when the patient is Homozygous.
In thalassaemias there is decrease in the quantity of normal globin chains in the hemoglobin. Thalassaemias are divided into the alpha thalassaemias, in which it is the production of alpha globin is deficient, and the beta thalassaemias, in which beta globin production is defective. The thalassaemias are divided clinically into Thalassaemia Minor and Thalassaemia Major, based on disease severity.

Beta-Thalassaemia
In this there is failure to synthesize beta globin chains. It is the most common type and is seen in highest frequency in the Mediterranean area. Those who are heterozygous, are thalassaemia minor, the condition is mild with mild anaemia and usually no disability. The homozygous are thalassaemia major, have profound anaemia after first 4 months of life.
Beta Thalassaemia major
Causes profound anaemia which has crippling effect on the health of the child. The child needs blood transfusion and chances for survival without transfusion for few years are low. Changes in the bone marrow (hyperplasia), results in head bossing and prominent malar eminences, which gives typical appearance of these diseases.
Development and growth of the child is retarded.
Folic acid deficiency develops.
Spleenomegaly (enlargement of spleen) is an early feature.
Cardiac enlargement with heart failure is common.
Repeated transfusions give rise to haemosiderosis (excessive deposition of iron ).
Beta Thalassaemia minor
Is detected often after treatment for mild anaemia with iron therapy fails. The condition is mild and often without any symptom.
Management of Beta Thalassaemia
- Blood transfusion to maintain haemoglobin.
- Bone marrow transplantation – Allogeneic bone marrow transplantation from HLA complatible sibling.
- Folic acid 5 mg daily.
- In case of iron overload – Desferrioxamine therapy.
- Splenectomy performed as late as possible.
Prevention
During early in pregnancy, DNA analysis of chorionic villus material can be done to identify a fetus with homozygous beta thalassaemia. Such pregnancy can be terminated. This examination can be done if both the parents are known to be carriers (thalassaemia minor).
Alpah Thalassaemia
It is due to reduction or absence of alpha chain synthesis. This type is common in the South East Asia. There are 2 alpha gene loci on chromosome 16 are therefore four alpha genes. If one is deleted there is no clinical effect. If two are deleted there may be a mild hypochromic anaemia. If three are deleted the patient has Hemoglobin H disease. If all four are deleted the baby is still born. Treatment of Hemoglobin H is similar to beta thalassaemia of intermediate severity.

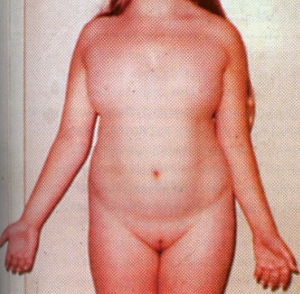 no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief.
no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief. The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.
The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.


