Malaria
Malaria is a type of fever which is a major cause of morbidity in the developing countries. Despite world-wide attempts to eradicate this disease it continues to be a big problem.
Caused by bite by female Anopheles mosquito which trasmits four types of parasites of genus Plasmodium – vivax, ovale, falciparum, malariae.
Plasmodium vivax is the most common and the Plasmodium falciparum is the most deadly.
Malaria is charactirised by relapsing fever with shaking chills, prostration, anaemia, and enlarged spleen.
Fever paroxysms are characterised by three phases:
Cold phase – lasting for 1-2 hours, during which sudden feeling of cold is followed by shivering, then intense rigors.
Hot phase – lasting for 3-4 hours, when there are hot flushes with headache and exhaustion as temperature reaches its peak.
Wet phase – lasting for 2-4 hours, when profuse sweating occurs as temperature returns to normal.
Fever paroxysms occur
every 48 hours in case of P.vivax, P.ovale,
every 72 hours in case of P. malariae.
In case of P.falciparum periodic fever paroxysms are not seen rather irregular intermittent fever or daily paroxysms may be seen.
Other symptoms – nausea, vomiting, cough, joint pains, abdominal and joint pain. Pallor and jaundice.
Death due to vivax malaria is very rare. Most malaria related fatalities occur due to P. falciparum malaria that can kill a non immune person within a week or two of infection.
Red Blood Cells which are invaded by the P.falciparum become very sticky and attach themselves to the walls of the capillaries. This blocks the microcirculation and thus causes cellular hypoxia, hypoglycemia, lacticacidosis and increased cellular permeability. These changes result in cerebral, pulmonary and renal manifestations.
Complications of Falciparum Malaria.
1. Cerebral Malaria – should be suspected in cases with history of short fever followed by deep unconsciousness in endemic areas. This accounts for 80% of deaths from acute malaria.
2. Algid Malaria – characterised by vomiting, nausea, diarrhoea, dehydration, low blood pressure, rapid respiration and low urine output.
3. Blackwater Fever – is caused by sudden extensive destruction of RBCs in the blood. Haemoglobin is found in the urine and this condition may precipitate renal failure. At times this condition is precipitated by the administration of Quinine to the patient.
4. Pernicious Anaemia – when blood contains a very high number of malaria parasite leading to rapid destruction of the RBCs.
5. Pulmonary Oedema and Adult Respiratory distress syndrome.
6. Vascular Collapse and Shock with Hypothermia and Adrenal Insufficiency.
7. Febrile Convulsions.
8. Hyperpyrexia
9. Metabolic Acidosis
In the management of Malaria the important and critical aspect is the diagnosis and treatment of falciparum malaria as soon as possible as deterioration of the patient’s condition can be rapid and sudden.
Besides drug treatment management of serious sign and symptoms are also very important.
Drug treatment of Malaria
Chloroquine: 600 mg immediately, 300 mg after 6 hrs., 300 mg on day 2, 300 mg on day 3 — for all four types of malaria.
Artemether: For Chloroquine resistant falciparum malaria.
day 1 – 80 mg IM twice.
Next 4 days 80 mg Once daily.
Quinine: For Chloroquine resistant falciparum malaria.
Slow infusion over 4 hrs. Initial loading dose 20 mg /kg followed every 8 hrs. with 10mg / kg. for 10 days.
Mefloquine: For Chloroquine resistant falciparum malaria.
750 mg immediately. 500 mg 8 hrs. later.
Primaquine: given after a course of chloroquine for the elimination of P. vivax and P. ovale from the body.
Other drugs: Pyrimethamine/sulfadoxine combination. Doxycycline.
Caution :
Quinine or Chloroquine not to be given as IV bolus.
Dexamethasone IV , Manitol, Heparine to be avoided in Cerebral Malaria
Prophylactic medication for Malaria.
In endemic areas regular intake of chloroquine 2 tabs. for adults or Pyrimethamine/sulfadoxine 1 tab. for adults weekly may be use to prevent malaria.


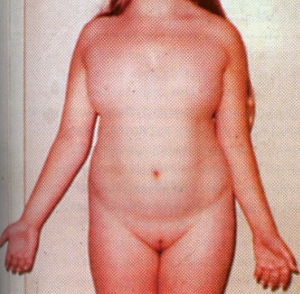 no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief.
no menses occur. In a small percentage of cases some menstruation may occur. Indeed, occasionally minimally affected women become pregnant; the reproductive life in such individuals is brief. The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.
The diagnosis is made either at birth because of the associated anomalies or at puberty when amenorrhea and failure of sexual development are noted.The external genitalia are of female type but remain immature and do not grow as in adult females. There is no breast development. Internally the fallopian tubes and uterus are also immature. On the both sides the ovaries are grossly underdeveloped.Skeleton and the connective tissue are also involved. Swelling of the hands and feet, webbing of the neck, low hairline, skin folds on the back of the neck, a shield like chest with widely spaced nipples, and growth retardation. These features suggest the diagnosis in infancy. Ears may be deformed. A fishlike mouth, Eyes may show ptosis – drooping of the upper eye lids.



